
Statistical analysis
IMPC data originates from many types of assays. Each assay has different characteristics so they cannot be analysed with a single statistical approach. As of data release 12, the IMPC uses a toolkit called OpenStats, available as an R package, to apply appropriate statistical methods for each data type. Previous releases relied on PhenStat for statistical analysis, also available as an R package here.
The OpenStats package provides statistical methods for the identification of abnormal phenotypes with an emphasis on high-throughput dataflows. The package contains:
- Dataset pre-processing and checking for minimal requirements; and
- 3 statistical tests to identify significant genotype effect on the phenotype:
- Fisher’s exact test for categorical data
- Linear mixed model (LMM) for continuous data
- Reference range plus (RR+) model for low N continuous data
All analysis frameworks output a statistical significance measure, an effect size measure, model diagnostics (when appropriate), and data visualization (particularly chart pages).
Further documentation on the OpenStats package can be found here, while the preprint paper can be accessed here.
Categorical data
Categorical data are analysed using Fisher’s exact test. This method involves constructing a contingency table to split animals up by phenotype and biological group, e.g. mutant versus baseline. Currently, phenotypes for a given trait are grouped together and compared to the frequency of normal phenotypes collectively. Examples of this type of analysis include abnormal coat colour and abnormal eye morphology.
Continuous data
Linear mixed model
Continuous data are analysed using a linear mixed model. This method uses a linear equation to model measurements as a function of genotype and additional variables including gender, weight, and batch (date of measurement). The batch is treated as a random effect except in the case of measurements that are taken over time (e.g. body weight), in which case it is included in the covariates. Interactions between variables such as genotype and sex can also be modelled.
This approach allows the statistical procedure to determine significant shifts that can be attributed to the genotype as opposed to the other covariates. Examples of this type of analysis include hypoactivity in open field and circulating insulin levels in clinical chemistry.
Relevant reading:
Impact of temporal variation on design and analysis of mouse knockout phenotyping studies
The fallacy of ratio correction to address confounding factors
Reference range plus
In cases where a linear mixed model fails, OpenStats offers an alternative analysis using the reference range plus model. This technique involves comparing measurements to predefined ranges that are deemed to capture acceptable variability in wildtypes. These comparisons, which lead to discrete values (within range vs. out-of-range), are then assessed statistically using a Fisher’s exact test.
Viability and fertility data
Viability and fertility data are treated separately with a custom approach. We refer to these as line level parameters. These data are collected and processed by the phenotyping centers, which use statistical methods appropriate for their breeding scheme. IMPReSS has specific information on how adult and embryo viability as well as fertility data are analysed.
Parallel coordinates viewer
Control selection strategy
One side effect of producing data in a high throughput pipeline is that the input data for a statistical calculation might be produced over multiple days. Environmental fluctuations have been identified as a confounding factor when comparing data gathered on different days. The IMPC describes this as a “batch effect” and it is treated as a random effect in the Mixed model framework.
Relevant reading:
Please, see this IMPC paper on a soft windowing application to improve analysis of high-throughput phenotyping data, Bioinformatics 36, 1492–1500 (2020)
The data sets to be analysed are identified using unique combinations of these fields:
| Field | Description |
| Background strain | The original strain from which the mutant specimen was derived. |
| Allele / Colony | The genomic variation in the mutant. The allele describes the character and the severity of the mutation, Genotype effect terms. |
| Zygosity | WT (wildtype or +/+) The wild type allele. Heterozygous (het) The mutation occurred in one copy of the allele. Homozygous (hom) The mutation occurred in both copies of the allele. Hemizygous (hemi) The mutation occurred in a sex-linked allele where, normally, only a single copy exists in the WT. |
| Pipeline | The standardized phenotyping pipeline as described in IMPReSS Pipelines. |
| Procedure | The standardised set of procedures (experiments) as described in IMPReSS procedures. |
| Parameter | The standardised set of measurements as described in IMPReSS parameters. |
| Metadata group | Some parameters are indicated as “procedureMetadata” type. Some of these metadata are used to group comparable data together as described on the IMPReSS parameters page under the “Required For Data Analysis” section. The parameters that are marked as “Required For Data Analysis” are collectively identified by an identifier called the metadata group. |
| Organisation | The phenotyping organisation that performed the experiment and collected the data. |
| Sex[1] | The sex of the specimens. When analyzed using the linear mixed model, males and females are analysed together to determine the Sex and Sex*Genotype interaction effect terms. [1] – optional |
Statistics to Phenotype
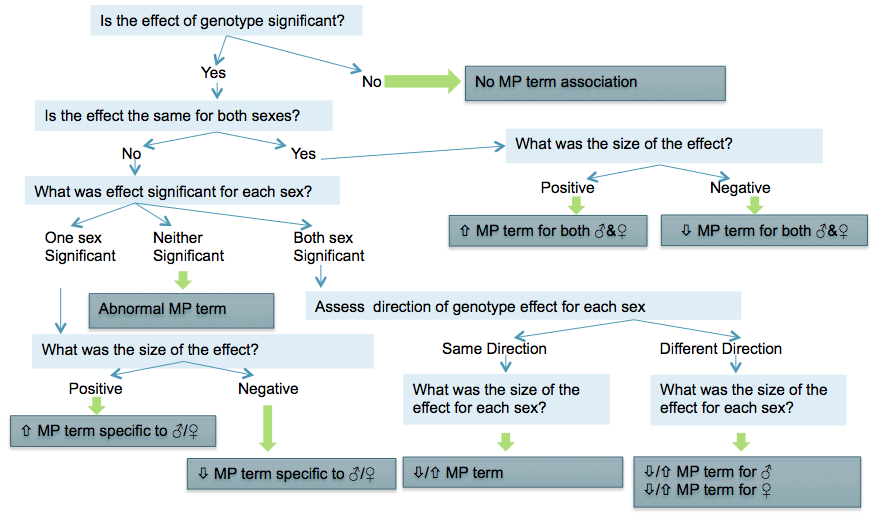
If the mutant genotype effect represents a significant change from the control group at the IMPC level of 0.0001 for continuous and categorical measurements, then the IMPC pipeline will attempt to associate a Mammalian Phenotype (MP) term to the data (Mammalian Phenotype Ontology).
The particular MP term(s) defined for a parameter are maintained in IMPReSS. Frequently, the term indicates an increase or decrease of the parameter measured, as compared to controls. If this is not appropriate, then an abnormal call may be indicated.
When a statistical result is determined as significant, the following diagram is used for associating MP terms:
Significance
When a mutant genotype effect P value is less than 1.0E-4 (i.e. 0.0001), it is considered significant.
Please see the section What P value does the IMPC use?